Sickle Cell Anemia
Sickle cell anemia is a congenital hemolytic anemia that occurs primarily but not exclusively in African-Americans. The condition results from a defective hemoglobin molecule (hemoglobin S) which causes red blood cells (RBCs) to roughen and become sickle-shaped. Such cells impair circulation, resulting in chronic ill health (fatigue, dyspnea on exertion, and swollen joints), periodic crises, long-term complications, and premature death. At present, only symptomatic treatment is available. Half of such patients die by their early 20s; few live to middle age.
Note: A technical writing student at the University of Texas at Austin wrote this definition in the 1980s. Technical accuracy cannot be guaranteed.
Causes and Incidence
Sickle cell anemia results from homozygous inheritance of the hemoglobin S-producing gene, which causes substitution of the amino acid valine for glutamic acid in the B hemoglobin chain. Heterozygous inheritance of this gene results in sickle cell trait, generally an asymptomatic condition. Sickle cell anemia is most common in tropical Africans and in persons of African descent. About 1 in 10 African-Americans carries the abnormal gene. If two such carriers have offspring, there is a 1 in 4 chance that each child will have the disease. Overall, 1 in every 400 to 600 African-American children has sickle cell anemia. This disease also occurs in Puerto Rico, Turkey, India, the Middle East, and the Mediterranean area. Possibly, the defective hemoglobin S-producing gene has persisted because in areas where malaria is endemic, the heterozygous sickle cell trait provides resistance to malaria and is actually beneficial.
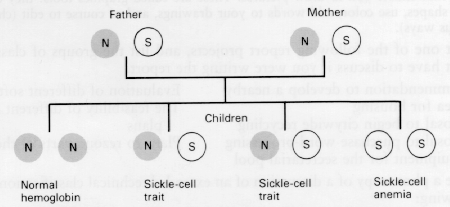
Passage of the sickle cell gene from parents to offspring
The abnormal hemoglobin S found in such patients' RBCs becomes insoluble whenever hypoxia occurs. As a result, these RBCs become rigid, rough, and elongated, forming a crescent or sickle shape. Such sickling can produce hemolysis (cell destruction). In addition, these altered cells tend to pile up in capillaries and smaller blood vessels, making the blood more viscous. Normal circulation is impaired, causing pain, tissue infarctions, and swelling. Such blockage causes anoxic changes that lead to further sickling and obstruction.
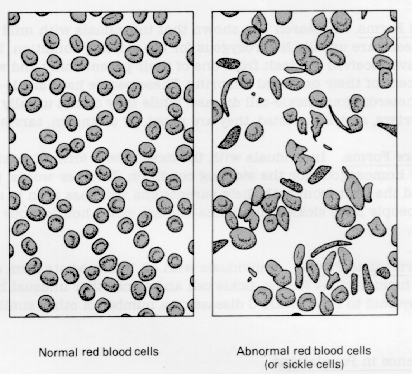
Normal and sickled red blood cells
Symptoms and Types of Sickle Cell Anemia
A number of symptoms are associated with sickle cell anemia, and in particular several types of crises.
Symptoms. Characteristically, sickle cell anemia produces tachycardia, cardiomegaly, systolic and diastolic murmurs, pulmonary infarctions (which may result in cor pulmonale), chronic fatigue, unexplained dyspnea or dyspnea on exertion, hepatomegaly, jaundice, pallor, joint swelling, aching bones, chest pains, ischemic leg ulcers (especially around the ankles), and increased susceptibility to infection. Such symptoms usually don't develop until after 6 months of age, since large amounts of fetal hemoglobin protect infants for the first few months after birth. Low socioeconomic status and related problems, such as poor nutrition and low educational levels, may delay diagnosis and supportive treatment.
Periodic crises. Infection, stress, dehydration, and conditions that provoke hypoxia—strenuous exercise, high altitude, unpressurized aircraft, cold, and vasoconstrictive drugs—may all provoke periodic crisis. Painful crisis. A painful crisis (vaso-occlusive crisis, infarctive crisis), the most common crisis and the hallmark of this disease, usually doesn't appear until age 5 but recurs periodically thereafter. It results from blood vessel obstruction by rigid, tangled sickle cells, which causes tissue anoxia and possibly necrosis. It is characterized by severe abdominal, thoracic, muscular, or bone pain and possibly increased jaundice, dark urine, or a low-grade fever. Autosplenectomy, in which splenic damage and scarring is so extensive that the spleen shrinks and becomes impalpable, occurs in patients with long-term disease. Such autosplenectomy can lead to increased susceptibility to Diplococcus pneumoniae sepsis, which can be fatal without prompt treatment. After the symptoms of crisis subside (in 4 days or several weeks), infection may develop, often indicated by lethargy, sleepiness, fever, or apathy.
Anaplastic crisis. Associated with infection is the anaplastic crisis (megaloblastic crisis) which results from bone marrow depression and is associated with infection, usually viral. It is characterized by pallor, lethargy, sleepiness, dyspnea, possible coma, markedly decreased bone marrow activity, and RBC hemolysis.
Acute sequestration crisis. In infants between 8 months and 2 years old, an acute sequestration crisis may cause sudden massive entrapment of red cells in the spleen and liver. This rare crisis causes lethargy and pallor, and if untreated, can progress hypovolemic shock and death. In fact, it's the most common cause of death in sickle cell children under 1 year.
Hemolytic crisis. Hemolytic crises are quite rare and usually occur in patients who have glucose-6-phosphate dehydrogenase (G-6-PD) deficiency with sickle cell anemia. It probably results from complications of sickle cell anemia, such as infection, rather than from the disorder itself. Hemolytic crisis causes liver congestion and hepatomegaly as a result of degenerative changes. It worsens chronic jaundice although increased jaundice doesn't always point to a hemolytic crisis.
Any of these crises are possible in sickle cell anemia patients with pale lips, tongue, palms, or nail beds; lethargy; listlessness; sleepiness, with difficulty awakening; irritability; severe pain; temperature over 104° F (40° C) or a fever of 100# F (38° C) that persists for 2 days.
Long-term complications. Sickle cell anemia also causes a number of long-term complications. Typically, children with sickle cell anemia are small for the age, and puberty is delayed. (However, fertility is not impaired.) If they reach adulthood, their bodies tend to be spiderlike—narrow shoulders and hips, long extremities, curved spine, barrel chest, and elongated skull. An adult usually has complications stemming from infarction of various organs, such as retinopathy and nephropathy. Premature death usually results from infection, or repeated occlusion of small blood vessels and consequent infarction or necrosis of major organs. For example, cerebral blood vessel occlusion causes cerebrovascular accident.
Diagnosis
A positive family history and typical clinical features suggest sickle cell anemia; a stained blood smear showing sickle cells, and hemoglobin electrophoresis showing hemoglobin S confirm it. Ideally, electrophoresis should be done on umbilical cord blood samples at birth, especially if the parents are known to carry the sickle cell trait. Additional lab studies show low RBC, elevated WBC and platelet count, decreased erythrocyte sedimentation rate (ESR), increased serum iron, decreased RBC survival, and reticulocytosis. Hemoglobin may be low or normal. During early childhood, palpation may reveal splenomegaly, but as the child grows older, the spleen shrinks and splenic function is impaired.
Treatment
Treatment is primarily symptomatic and can usually take place at home. If the patient's hemoglobin drops suddenly, as in an anaplastic crisis, or if his condition deteriorates rapidly, hospitalization is needed for transfusion of packed red cells. In a sequestration crisis, treatment may include blood transfusion, oxygen administration, and large amounts of oral or IV fluids. So far, research to find an effective antisickling agent hasn't been successful.
I would appreciate your thoughts, reactions, criticism regarding this chapter: response—David McMurrey